PhylomeDB User's Manual Index
Phylomes represent the complete collection of evolutionary histories for each gene encoded in a genome. Reconstruction of such large amount of trees has become feasible in recent years thanks to the increase in computation power and improvements in the reconstruction methods. It is now possible to generate those phylomes in an automatic way, generating thus large amounts of data that need to be stored. PhylomeDB was build in order to store all the data derived from phylome reconstruction. As such it now includes hundreds of thousands of trees and alignments. PhylomeDB has also been complemented in recent years with mappings to other external databases such as UniProt, Ensembl or Genolevures. Orthology and paralogy predictions have been extended to include metaPhOrs predictions (http://orthology.phylomedb.org/), which provides a consensus prediction of evolutionary relationships based not only on one tree but on multiple trees found across numerous databases (PhylomeDB, EnsemblCompara, EggNOG, OrthoMCL, COG, Fungal Orthogroups, and TreeFam). In this last version, phylomeDB has enhanced the tree visualization module in order to show protein domains that were mapped onto the tree sequences. All the data can be downloaded either from the tree page, where information of a particular tree can be obtained, or through our ftp page (ftp://phylomedb.org/phylomedb/) for larger datasets.
User Guide
Phylomes are the complete collection of phylogenetic trees derived from a completely sequenced genome. In order to reconstruct them an automatic pipeline was designed that replicates a standard tree reconstruction process. For detailed information about the pipeline used in phylomeDB check the pipeline information page here. Each phylome is formed by a seed proteome, which is the starting point of phylome reconstruction, and a group of proteomes against which the seed is queried. PhylomeDB has been designed to store all the data generated during a phylome reconstruction, including phylogenetic trees, alignments and orthology predictions.
Starting Page
The image below shows the main page of phylomeDB and indicates the most important features so you can quickly start using phylomeDB.
a) phylomeDB abstract: Here you can find a brief introduction about what phylomeDB is and how phylomeDB works.
b) Content browser: Using these links you can access other parts of phylomeDB: Home: Go back to the home page whenever you want. Search: This option will open the possibility to search for different items in PhylomeDB. All Phylomes: This will link you to the complete list of publicly available phylomes. Downloads: Leads to a ftp page where information associated to the phylomes can be downloaded. Downloads from single trees or alignments can be done directly from the tree page. Help: leads to this manual. FAQ: Frequently asked questions. About: Information about how to cite phylomeDB and about the people involved in maintaining the database.
c) News: Latest news involving phylomeDB
d) Twitter: Breaking news about the phylomeDB world (collaborations, new papers, new phylomes...). Follow/tag with the @phylomedb handle.
e) Who are we?: Learn more about us and the latest work from our group.
f) Where are we?: Logos of our institutions or funding bodies.
The phylome pipeline mimics the steps a phylogeneticist would follow to reconstruct a gene phylogeny: homology search, multiple sequence alignment and tree reconstruction. Small variations can be found across phylomes depending on the year they were reconstructed or the particular needs of each phylome. A detailed description of the methodology used in each phylome can be found in the main page of each specific phylome (i.e.: http://phylomedb.org/phylome_533). Here we provide a general overview of the steps taken in phylome reconstruction, phylomes reconstructed after the beginning of 2019 have undergone small modifications which will be detailed below.
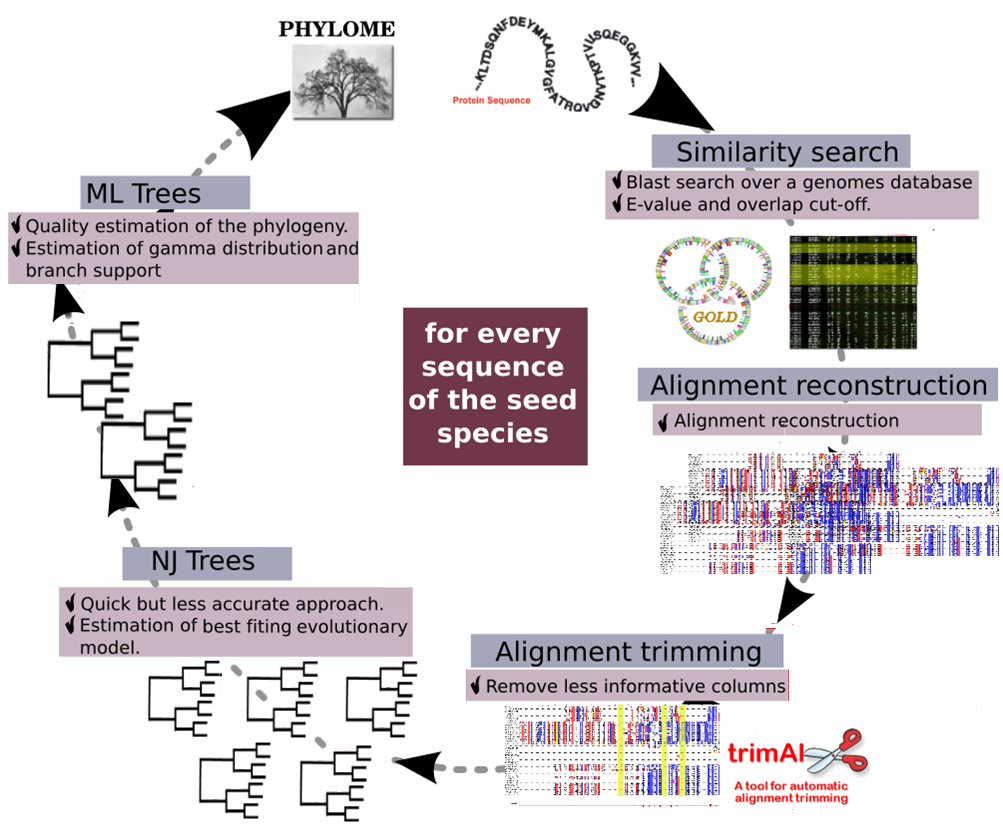
Data collection:
Each phylome is reconstructed with a set number of proteomes. These proteomes have been downloaded from different databases or can belong to collaborating sequencing projects. The details of the source of each proteome can be found in the main phylome page. Take into account that one single species can be represented by a different proteome version in two different phylomes. Phylomes are reconstructed starting from one of the proteomes, which is called the seed proteome. This will be the proteome that will be fully represented in the phylome, while the other proteomes will only appear in trees when they have homologous sequences to the seed. In some instances meta-phylomes are reconstructed. Those are groups of phylomes that use the same set of proteomes and start using different seed proteomes.
Homology search:
For each protein encoded in the seed proteome a Smith-Waterman search is performed against the database that comprises the selected proteomes to retrieve a set of proteins with a significant similarity. Results are filtered based on their e-value and the percentage of overlap between the query sequence and the hit. A limit in the number of accepted hits is also used.
Multiple Sequence Alignments (MSA):
In the older phylome versions MSA were performed with MUSCLE and then regions with a high amount of gaps were removed using trimAl. In the last years this step has been modified to adapt to a more robust pipeline. Now sets of homologous protein sequences are aligned using three different alignment programs chosen among: MUSCLE, MAFFT, DIALIGN-TX and KALIGN. Alignments are performed in forward and reverse direction and the six resulting alignments are combined using M-COFFEE. The resulting alignment is then trimmed using trimAl using a consistency cutoff of 0.1667 and a gap score cutoff of 0.1.
Phylogenetic reconstruction:
Maximum likelihood (ML) trees are reconstructed for each seed protein that has at least two homologs. One of the important steps in ML reconstruction is to choose the correct evolutionary model. In earlier phylomes we opted to reconstruct trees with different models (usually JTT, WAG, Blossum62 and VT) and then chose the one with the best likelihood according to the Akaike Information Criterion (AIC). Until 2019 the approach consisted in selecting the best model from a collection of Neighbor Joining trees that were reconstructed using scoredist distances as implemented in BioNJ. The likelihood of the topology was computed, allowing branch-length optimisation, using seven different models (JTT, LG, WAG, Blosum62, MtREV, VT and Dayhoff), as implemented in PhyML. The evolutionary models best fitting the data were determined by comparing the likelihood of the used models according to the Akaike Information Criterion (AIC). Maximum likelihood trees are then derived using the selected models. In all cases ML trees were reconstructed using a discrete gamma-distribution model with four rate categories plus invariant positions, the gamma parameter and the fraction of invariant positions were estimated from the data. As of 2019, model selection and maximum likelihood tree reconstruction have been migrated to IQTREE. To optimize time only a number of models are considered by default (DCmut, JTTDCMut, LG, WAG, VT), though that may be adapted for a particular phylome. Categories for FreeRate model were set to range between 4 and 10. The best model according to the BIC criterion was selected. 1000 rapid boostraps were calculated.
Phylome details:
While all the phylomes follow a similar pipeline, each one has unique features such as the proteomes involved in the reconstruction or small changes in the pipeline. All the information pertaining to how a phylome was reconstructed can be found in the main phylome page. To access it, you can go to the "All phylomes" link on the top of the page. This will lead you to the complete list of phylomes that are currently available. Clicking on the phylome name will take you to the main phylome page. The phylome page is currently composed of four sections:
a) Description. Here are all the details on how the phylome was reconstructed including which programs were used and their version, which relevant parameters were used and how many evolutionary models were used to reconstruct the trees.

b) Gene trees. Shows a list of gene trees for the phylome. It is similar to the previous browse feature in v4 of phylomedb.

c) Species and taxonomy. One or several images of the species tree enclosing all the proteomes used in the phylome are provided in the phylome page. This information will give a visual evolutionary context for the phylome.

d) Proteomes. Information about the proteomes used in this phylome can be found here. The information found in the table includes: taxonomy ID of the species, species name, which is linked to its page in NCBI taxonomy, the proteome version, the source and the date in which it was included. The proteome version is formed by two fields. The first is the species assigned code, which is normally composed of 5 characters and should match the UniProt mnemonic assignation. In cases in which UniProt has no mnemonic code assigned, the taxID is used. In extreme cases where a species does not have a taxID, a temporal taxID is given. The second parameter refers to the version of the proteome used in this phylome. So different human phylomes can use different human proteomes.

Orthology prediction
PhylomeDB uses a phylogeny-based algorithm to detect duplication and speciation events on the trees. This algorithm is called species-overlap and is described in more detail in Huerta-Cepas et. al (2007). In contrast to standard phylogeny-based methods that use reconciliation of the gene tree with a given species tree to infer duplication events, our approach does not require any previous fully resolved species topology, as far as the trees are rooted. The orthology prediction algorithm is run independently for each seed protein using the corresponding phylogenetic tree. To establish orthology and paralogy relationships, the algorithm goes over all the nodes in the tree deciding whether they are a duplication and a speciation node. For each node two tree partitions are defined that contain the sequences connected to each of the two children nodes. Then, a species-overlap score is defined between the two partitions as follows: species common to both partitions/species in any of the partitions. Finally, if the score is higher than a given threshold the node is mapped as a duplication event, otherwise it is considered a speciation event. So far in all phylomes the species-overlap threshold was set to 0.0 - that is, no common species between the two partitions were allowed - because this produced the best results in the benchmark, as explained in the Human phylome paper. Once all the nodes in the tree are marked as a duplication or speciation event, the algorithm establishes orthology relationships between the seed protein and other proteins in the tree. For each protein, the algorithm tracks the nodes that connect it to the seed protein and establishes an orthology relationship only if this connection proceeds exclusively through speciation nodes, disregarding intra-specific duplications. After mapping speciation and duplication nodes onto the phylogeny, several situations may arise in which orthology relationships are not one-to-one relationships, but rather one-to-many or many-to-many.

All the duplications and speciation events are mapped onto each phylogenetic tree in the form of different coloring. Blue nodes stand for speciation nodes while red nodes represent duplication nodes.
The tree interface is the central feature of phylomeDB. The tree is fully interactive and can be manipulated as well as used in order to obtain additional information. Clicking on different parts of the tree will produce drop-down menus through which a tree can be manipulated. The default tree shown after a search belongs to the most recently reconstructed phylome and the best evolutionary model within this phylome. You can change the tree selection using the menu placed just on top of the tree.
Main menu
The features explained below will allow you to select which tree you want to visualize, how you want to visualize it and will allow you to download the data.

a) This will provide the list of trees where your protein of interest has been used as seed. There is the possibility that this list is empty. In this case the protein has never been used as seed.
b)This option will allow you to switch evolutionary models that were used in the phylome. When building trees during a phylome reconstruction, up to seven evolutionary models are tested. Maximum likelihood trees for only a few (1 or 2) of them are reconstructed and shown in phylomeDB.
c) We call collateral trees those trees in which the sequence of interest appears but has not been used as seed. This list of trees can be found here. If the sequence was never used as seed, the tree shown in the tree page will be a collateral tree.
d) This row of tools will allow you to manipulate the tree image and download data. The first two tabs will open additional menus. The first one pertains to the annotations shown in the tree. Those annotations that are checked are currently shown in the tree image, such as pfam domains or the species name. If you want to show an additional feature, you can just mark it on the menu and click on Refresh. Not all leaves in the tree may have all the features, for instance genomes that were not taken from Ensembl will not have an ensembl annotation. The second menu is a search menu, it will allow you to search for leaves in the tree that fulfil a particular requirement such as species name or taxonomic group. The remaining tabs will allow you to download the data specific for the tree or to see the alignments.
Tree image
The tree image can be modified using different commands that are encoded in menus shown when clicking on the internal nodes or leaves of the tree.a)The tree image in itself encodes several features that may be of interest for the user.
By default the leaves will be shown by the code they were initially integrated into phylomeDB. Next to them the name of the species they belong to will be also displayed. The nodes are coloured depending on whether they are speciation (blue) or duplication (red) nodes. Also, for each tree, an aLRT support is calculated, if this support is lower than a given threshold the number will be shown and a bubble will be seen around the node. The bigger the bubble, the more unstable the node is.

b) Clicking on an internal node will show the menu found in b. This allows small manipulations in the tree such as swapping the order of the children in the tree or setting the root at this point. Information of the taxonomic age of the node can be also found here. This age is calculating by seeing which taxonomic group is the common ancestor of all the leaves derived from this node. The branch length and support are also displayed on the top of the menu.
c) Clicking on a leave will give similar options for modifying the tree but will also offer additional information pertaining to the leaf itself. So, for instance, the complete lineage of the species can be found there as well as other annotations (uniprot or ensembl codes, internal phylomeDB code, etc...). The lower part of the menu will allow you to access the sequence page from phylomeDB as well as link to external databases such as UniProt, Ensembl, NCBI Taxonomy or metaPhOrs.
d) and e) Leaves can be marked with two kinds of bubbles: a green bubble can represent the protein that was used as seed to reconstruct this particular tree or the protein you were looking for. A yellow bubble marks the results of a search.
Additional features
Next to the tree additional features can be marked. Right now there are two features implemented: Taxonomic range and protein domains.

a)Taxonomic range:This marks in different colors the different taxonomic groups to which a species or group of species belong. A leaf can belong to different taxonomic ranges of different levels (order, sub-order, family...). This can give an idea on how well the tree conforms towards the NCBI taxonomic classification.
b) Protein domains mapped according to Pfam can be found here. Each line represents the protein sequence where domains are represented in different shapes and colours while sequence regions without domain are colour-coded according to the amino-acids encoding the sequence. Straight lines represent gaps in the alignments.
Phylo Explorer
"Phylo Explorer" is an interactive visualization tool, that allow the users to browse the phylomes in our database and have a better view of the relations between the species and the phylomes they are in.
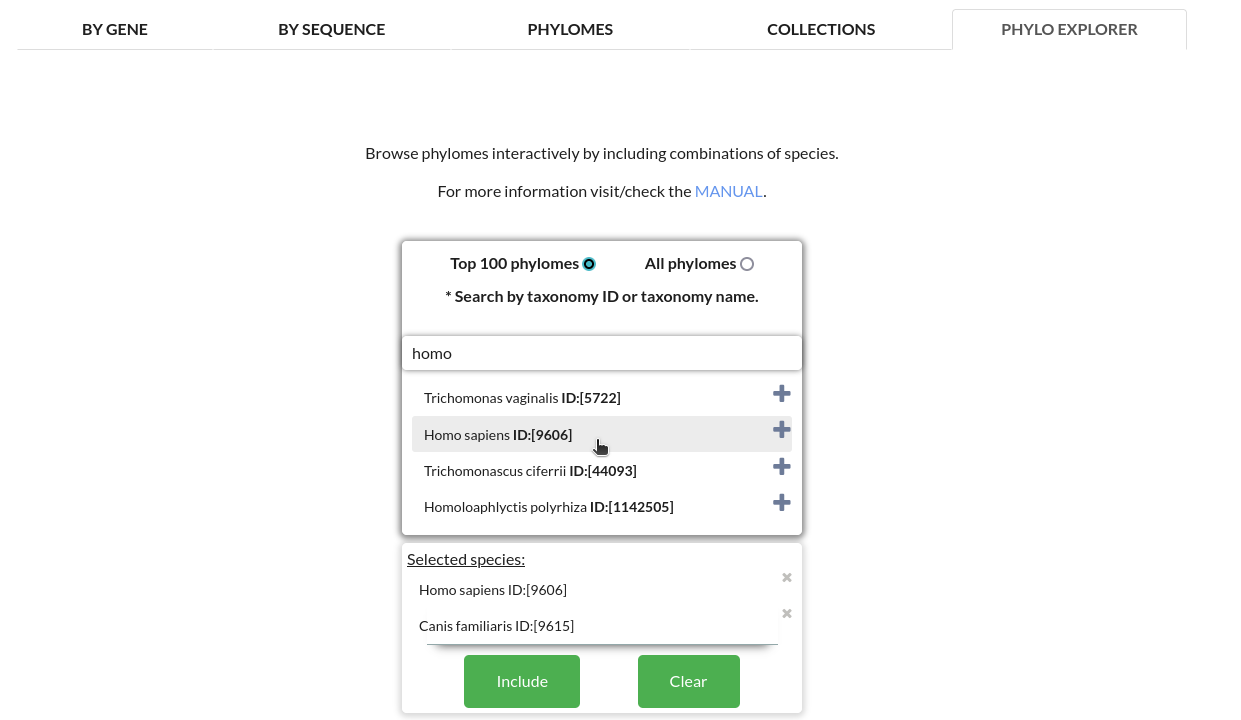
Selecting the species to be included in the explorer:
Searching and selecting by taxonomy names or taxonomy identifiers, species can be explicitly included in the generation of the explorer.
*Only phylomes which contain all the selected species will be included.
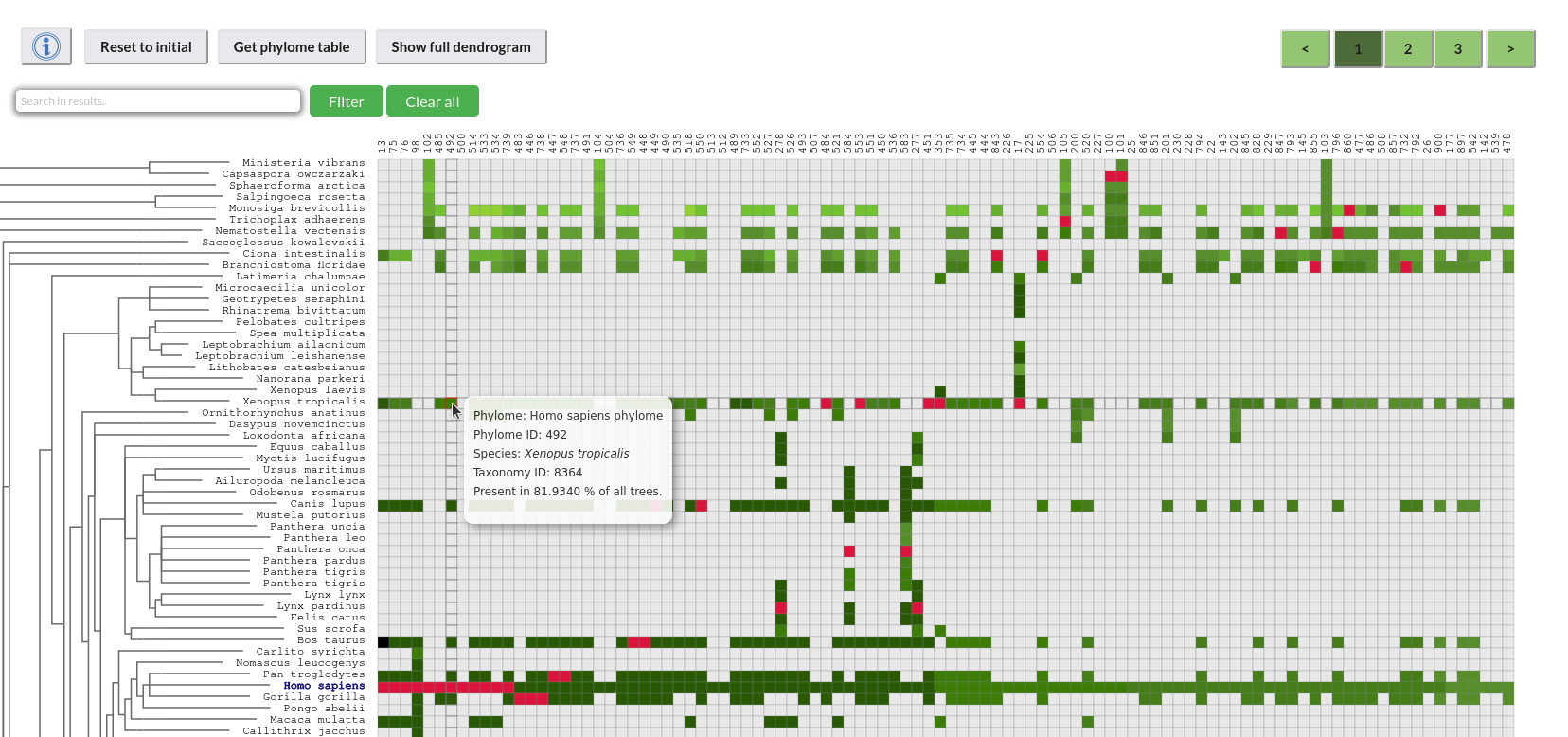
Description of the explorer's components:
Each column represents a phylome and each row a species. The color of a cell in the cross section indicates if the phylome of that column in what percentage contains that species in all of its trees.
- The dendrogram on the left side of the heatmap represents a taxonomy tree of the included species, corresponding to the relationship of their lineages.
- Columns are labeled with the identifiers of the included phylomes.
- Rows are labeled with the taxonomy names of the included species.


Meaning of the colors in the heatmap:
- Each shade of the green color increments by each 10th percentage value of the species presence in all the phylome's trees.
- Grey means that the phylome does not contain the species.
- Black means that the species is present in all of the phylome's trees.
- Red means that the species is the seed of the phylome.
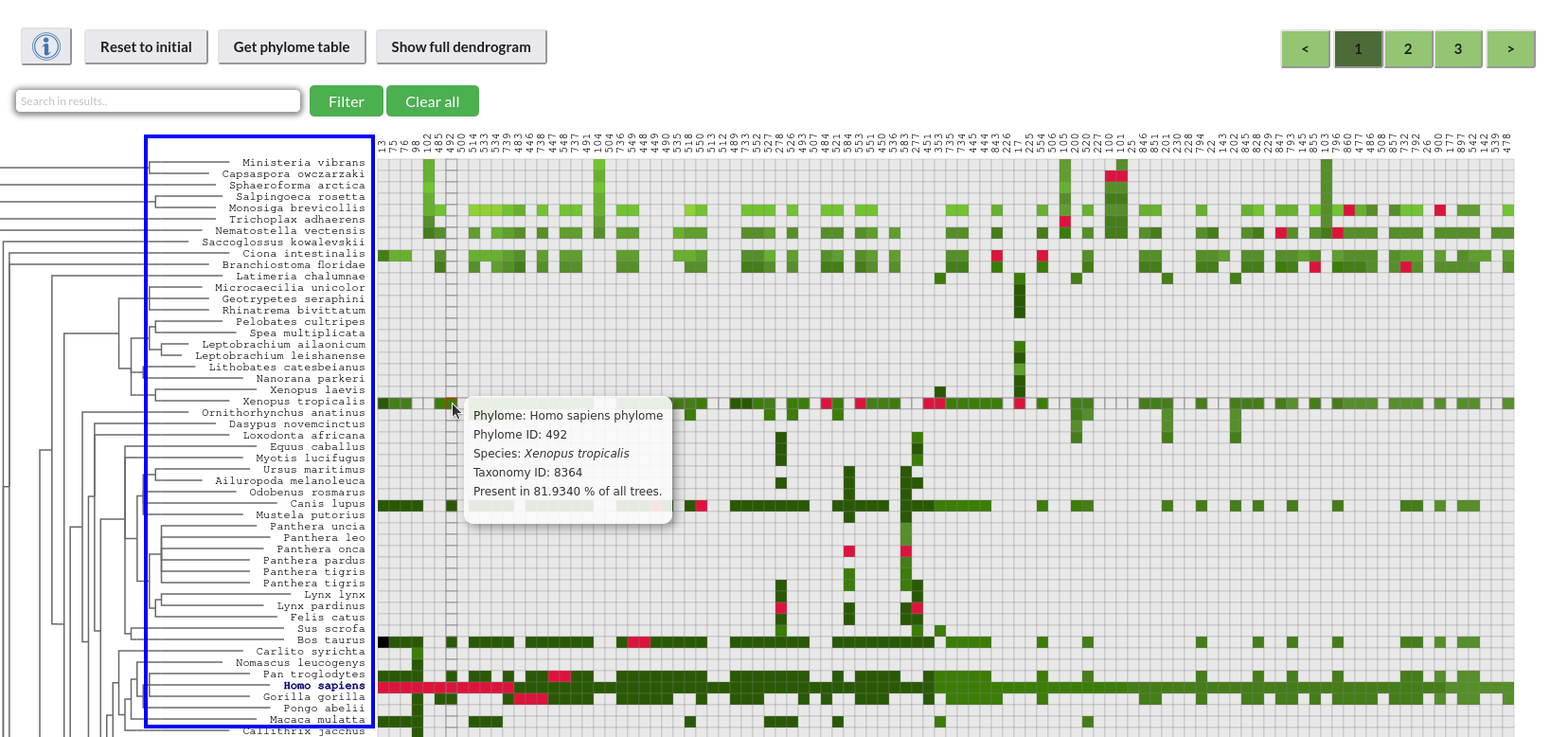
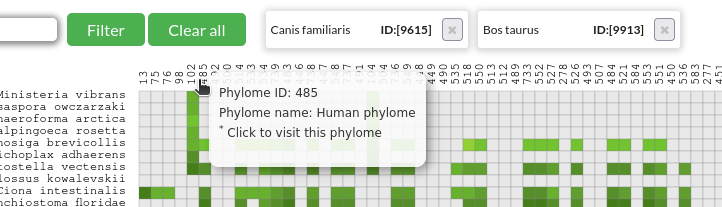
Narrowing down the explorer to certain species:
To reduce the explorer's content for a specific combination of species after the first inclusion, the filter bar can be used (first search bar directly above the dendrogram) to search between the included species and select them. After the selection, to initialize the filtering, click on the "Filter" button.

Selecting the species of interest to search:
By clicking on the cells or the dendrogram labels, species can be selected to get a data table of those phylomes, which include the combination of the selected species in all their trees.
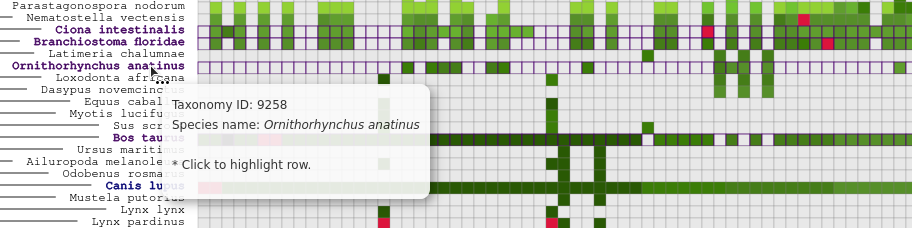
*Once the species are marked, click on the "Get phylome table" button.

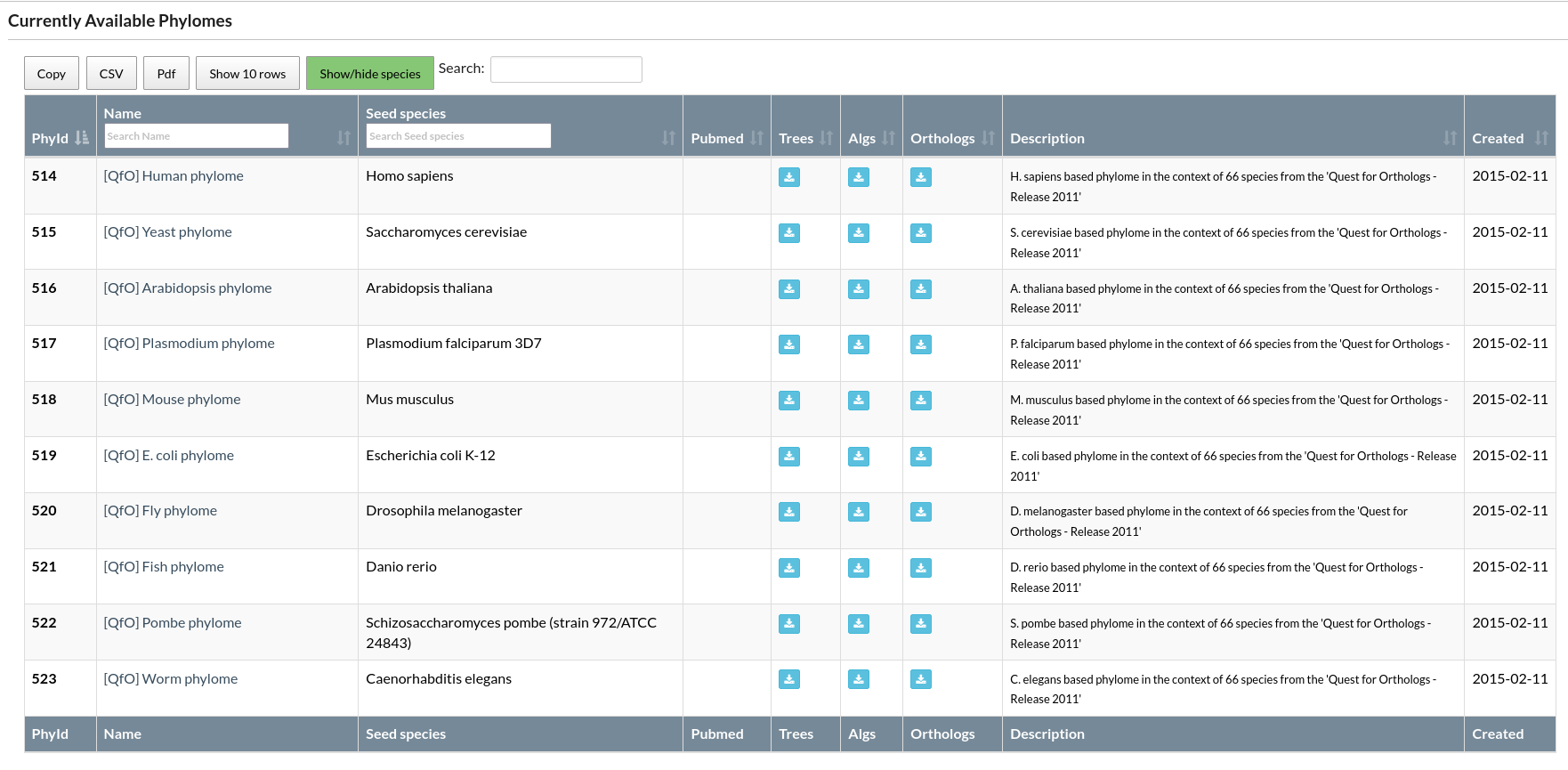
Checking out phylomes:
PhylomeDB has the option to browse phylomes as a data table ("Search" -> "Phylomes"), but a phylome's content can be accessed by clicking on the explorer's column labels.
Other pages
On the main browser of PhylomeDB several general options can be found.
Help: The Help tab links directly to the user manual which has been written to help new users understand how PhylomeDB works and the best way in which to use it.
FAQ: The Frequently Asked Questions comprise all the questions we believe a user may have when trying to use phylomeDB and their answers. This part is updated as new questions come up.
About: The last tab offers information on the PhylomeDB: members, contact information, how to cite phylomeDB, etc